
Introduction
Colorectal cancer (CRC) is a leading cause of cancer-related mortality worldwide, accounting for almost 10% of deaths.¹ The incidence of CRC is rising due to population aging and various dietary, lifestyle, and environmental factors.¹ Genetic factors, including mutations in tumor suppressor genes and oncogenes, play a crucial role in CRC pathophysiology and often drive changes in gene expression.² Studying these transcriptomic changes provides insights into the molecular mechanisms underlying CRC and enables the identification of differentially expressed genes, which can serve as biomarkers for diagnosis, prognosis, and treatment response.³
Re-analyzing public RNA-seq data offers an invaluable resource for such studies, allowing researchers to validate findings and discover novel insights. However, acquiring the data, running a pipeline at scale, and creating visualizations to derive insights can be challenging. Accessing large public datasets requires efficient data management and substantial computational resources.⁴ Running bioinformatics pipelines involves integrating various tools and ensuring reproducibility and scalability, which can be complex and time-consuming. Additionally, creating meaningful visualizations that accurately represent the data and uncover significant patterns necessitates advanced skills and tools.⁵
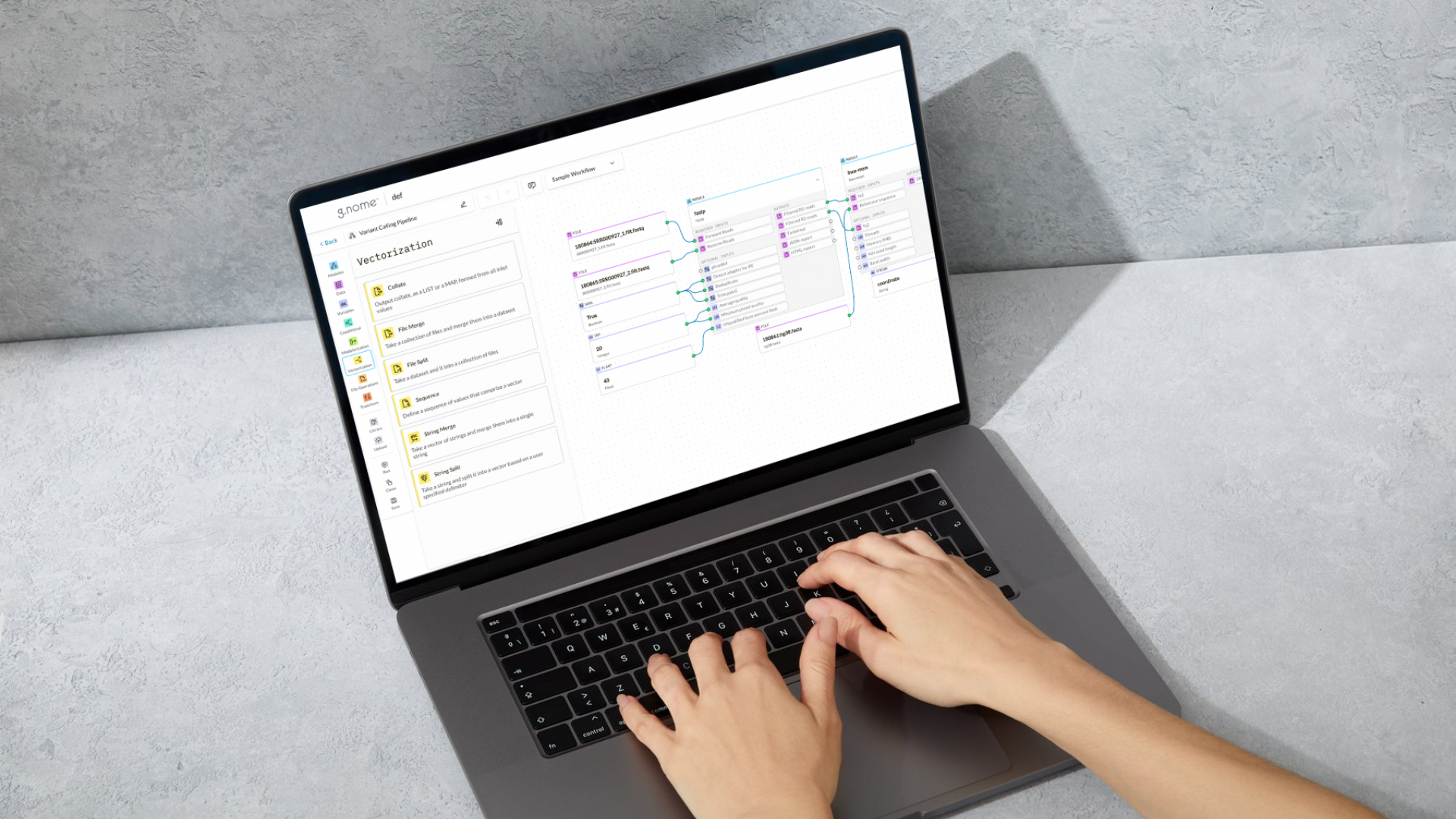
The Almaden Genomics g.nome® platform makes it easier to manage and analyze large RNA-seq datasets. g.nome streamlines the entire end-to-end process, from data acquisition to visualization, helping researchers quickly uncover valuable insights into gene expression patterns. To demonstrate this, we conducted a differential gene expression analysis on RNA-seq data from CRC patients, re-analyzing four transcriptomics datasets from the Sequence Read Archive (SRA) referenced in the study by Hosseini & Nemati.5 These datasets included samples of colorectal tumor tissues and adjacent normal tissues, providing a rich resource for understanding the molecular mechanisms of CRC. This re-analysis highlights the value of public data in validating findings and discovering new insights.
Objectives
- Demonstrate the effectiveness of the Almaden Genomics' g.nome platform in managing and analyzing large RNA-seq datasets.
- Illustrate the streamlined end-to-end process from data acquisition to visualization using g.nome.
- Conduct a differential gene expression analysis on CRC patient RNA-seq data.
- Validate findings and discover novel insights by re-analyzing publicly available transcriptomics datasets.
- Highlight the value of public data in understanding the molecular mechanisms of CRC.
Methods
We conducted a differential gene expression analysis on RNA-seq data from CRC patients, re-analyzing four transcriptomics datasets from the Sequence Read Archive (SRA) referenced in the study by Hosseini & Nemati5. The datasets included samples of colorectal tumor tissues and adjacent normal tissues (Table 1).
- Fetching RNA-seq Data: Using g.nome's workflow, we fetched RNA-seq datasets from the SRA database by inputting the corresponding accession numbers (Figure 1).
- Quality Control and Alignment: The data quality was assessed using fastqc, and reads were pre-processed with fastp. The STAR alignment algorithm was used to align filtered reads against the human genome reference sequence (Figure 4).
- Differential Expression Analysis: High-quality alignments were inputted into a workflow for differential expression analysis, where the featureCounts algorithm quantified RNA transcripts, and DeSEQ2 identified differentially expressed genes (Figure 5).
Background Research
Differential expression analysis helps to uncover molecular mechanisms underlying cancer progression by revealing genes that are significantly upregulated or downregulated in cancerous tissues compared to normal tissues. We conducted differential analysis of gene expression between tumor and adjacent normal tissues of CRC patients, based on the four transcriptomics datasets from the Sequence Read Archive (SRA) used in the recent publication of Hosseini & Nemati5 (Table 1).
|
SRA Accession Number |
# of Colorectal Tumor Tissues |
# of Adjacent Normal Tissues |
Study Name |
|
SRP219837 |
5 |
5 |
Epigenomics landscape of colorectal cancer |
|
SRP301216 |
5 |
6 |
Identification of genes associated with the onset of colorectal cancer by transcriptomic analyses of the adenoma-carcinoma sequence |
|
SRP344867 |
5 |
4 |
RNA Sequencing in Adenoma-Cancer Transition |
|
SRP245232 |
3 |
3 |
RNA-seq of CRC tissues |
|
Table 1. RNA-seq datasets used in this study. |
Using the g.nome Guided Workflow to Download Data

| Figure 1. Selecting data for the guided workflow in g.nome. |
The g.nome platform provides a user-friendly, guided workflow to simplify the process of downloading data for bioinformatics analysis. Here we see the setup for executing the nf-core fetchngs workflow. This workflow facilitates the efficient fetching of NGS (Next-Generation Sequencing) data from public repositories.
- Select Workflow: Start by selecting the desired workflow from the curated list.
- Label Run: Assign a label to the run to help track and identify the specific task.
- Execution: Click "Next" to proceed with the execution of the workflow. The platform will guide you through the subsequent steps, ensuring that all necessary parameters and inputs are correctly configured.
By using g.nome's guided workflow, researchers can streamline data acquisition processes, reduce setup time, and minimize errors, making it easier to manage large-scale RNA-seq datasets for further analysis.
Using g.nome for Bulk RNA-seq Data Analysis

| Figure 2. Using the guided workflow to process the data. |
Here we demonstrate how to use the g.nome platform for Bulk RNA-seq data analysis. The step-by-step process includes:
- Starting the Workflow: Begin by selecting the appropriate workflow for Bulk RNA-seq analysis. The guided interface helps users configure the workflow settings efficiently.
- Data Input: Upload your RNA-seq data files or specify the data source. The platform supports various data formats and sources, ensuring compatibility and ease of use.
- Parameter Settings: Adjust the analysis parameters as needed. The platform provides default settings optimized for common use cases, but users can customize these based on their specific requirements.
- Running the Analysis: Once all settings are configured, initiate the workflow. The platform will process the data and perform the analysis, providing real-time updates on the progress.
Using the g.nome platform, researchers can efficiently conduct comprehensive RNA-seq data analyses, from data acquisition to visualization, thereby accelerating the discovery of new insights in genomic research.
Differential Expression Analysis Visualization
| Figure 3. MA plots, heatmaps and volcano plots are among the visualizations used for differential expression analysis. |
The g.nome platform provides several key visualizations to help interpret the results of differential expression analysis:
- Differential Expression Report: Detailed reports on the differential expression analysis, including insights into upregulated and downregulated genes, their fold changes, and statistical significance. This report includes volcano plots, heatmaps, MA plots and Principal Component Analysis (PCA) plots.
- Differential Expression Results (DEseq2): The DEseq2 analysis identified differentially expressed genes between colorectal cancer (CRC) tissues and normal adjacent tissues, highlighting genes with significant changes in expression levels.
- Gene Count Data: The gene count matrix generated from the RNA-seq data quantifies the abundance of RNA transcripts across different samples.
- Gene Set Enrichment Results: Results of gene set enrichment analysis, identifying biological pathways and processes significantly enriched with differentially expressed genes, providing insights into the functional implications of gene expression changes.
- Pathway Analysis Results: Shows the enrichment of differentially expressed genes in various biological pathways, helping to understand the broader biological context of the gene expression changes.
- MultiQC Reports: MultiQC aggregates results from quality control tools like FASTQC and alignment tools, providing comprehensive reports on sequence quality and alignment metrics.
Results
Pre-Processing and Quality Control
Pre-processing of read files was performed using fastp. According to fastp, approximately 95% and 90% of bases in the sequencing run SRR16832117 had base quality values better than 30 and 20, respectively. fastp reports the number of reads that passed the quality filter as well as the number of reads that failed due to low quality, too many unresolved bases, and insufficient length. It also reports the number of reads with adapters trimmed as well as the number of bases trimmed due to adapters.

Figure 4. Read quality assessment by FASTQC. The graph shows per base sequence quality for forward reads corresponding to the sequencing run SRR16832117 from the study of Orouji et al.12
Alignment
Filtered direct and reverse reads were aligned against the pre-indexed reference sequence of the human genome using the STAR spliced read alignment algorithm. The alignments are stored in the widely used BAM (Binary Alignment Map) format. Using SRR16832117 as an example, there were 39 million reads with an average length of 294, of which 92% could be uniquely mapped to the genome sequence, 4.6% were mapped to multiple loci, and the remaining reads could not be mapped due to various reasons. In the final processing step, g.nome uses Picard tools to verify mate-pair information between reads and to mark duplicates originating from a single fragment of DNA.
Differential Expression Analysis
The dataset of high-quality alignments stored in BAM files serves as input to the main workflow for differential expression analysis. Note that the pre-processing and alignment workflow is used here as a sub-workflow. This illustrates the useful feature of g.nome, which allows combining simple sub-workflows into more complex processing pipelines hierarchically and reusing specific workflows in different contexts. The featureCounts algorithm quantifies the abundance of RNA transcripts in sequencing data by assigning reads to genes based on their alignment to the human genome. The resulting gene count matrix is used by DESeq2 to compare gene expression levels across different experimental conditions or groups to identify genes that are differentially expressed.
Key metrics include log fold change (indicating the magnitude and direction of change in gene expression) and p-value (measuring the statistical significance of the observed changes). The top 10 differentially expressed genes identified in this analysis are visualized in the volcano plot below. These genes have known roles in CRC, such as KRT23, which promotes CRC cell proliferation, and ETV4, which enhances tumor progression and metastasis.

|
Figure 5. Volcano plot visualizing the genes differentially expressed between CRC samples and normal adjacent tissues. The top ten differentially expressed genes listed in Table 2 are highlighted by larger circles. |
Top 10 Differentially Expressed Genes
|
Gene Name |
Log2 Fold Change |
Adjusted p-value |
Protein Name |
Known Roles in Colorectal Cancer |
|
KRT23 |
6.7 |
6.9e-23 |
Keratin, type I |
Promotes CRC growth by activating human telomerase reverse transcriptase, associated with CRC cell proliferation and migration. [6] |
|
ETV4 |
4.0 |
1.7e-22 |
ETS translocation variant 4 |
Enhances CRC cell proliferation, invasion, and metastasis, influences the tumor microenvironment. [7, 8] |
|
CPNE7 |
4.6 |
8.0e-22 |
Copine-7 |
Suggested as a prognostic factor and therapeutic target in CRC. [9] |
|
GRIN2D |
4.1 |
6.4e-21 |
Glutamate receptor ionotropic |
Angiogenic tumor endothelial marker specific to CRC vessels, correlated with improved survival in CRC patients. [10] |
|
BEST4 |
6.1 |
3.1e-20 |
Bestrophin-4 |
Suppresses epithelial-to-mesenchymal transition in CRC cells. [11] |
|
STRA6 |
4.5 |
6.6e-19 |
Receptor for retinol uptake |
Plays a key role in colon cancer stem cell maintenance and contributes to high-fat diet-induced colon carcinogenesis. [12] |
|
CDH3 |
5.8 |
2.0e-18 |
Cadherin-3 |
Elevated expression in CRC, demethylation linked to advanced CRC. [13, 14] |
|
KLK6 |
9.5 |
1.9e-17 |
Kallikrein-6 |
Prognostic biomarker due to dramatic upregulation in CRC, key role in colon cancer cell migration and invasiveness. [15, 16] |
|
LARGE2 |
3.7 |
3.1e-17 |
Xylosyl- and glucuronyltransferase |
Increased levels in CRC compared to benign colonic epithelial cells, involved in CRC cell migration and adhesion. [17] |
|
MMP7 |
7.5 |
1.2e-16 |
Matrilysin |
Required for CRC tumor formation, affects drug resistance. [18, 19] |
| Table 2. Top 10 genes differentially expressed between tumor and normal tissues |
Conclusions
Re-analyzing public RNA-seq data using the g.nome platform demonstrates the potential of leveraging existing datasets to gain new insights into cancer biology. This approach not only validates previous findings but also allows for the discovery of novel biomarkers and therapeutic targets. The g.nome platform's ease of use and robust analytical capabilities make it an invaluable tool for researchers aiming to perform complex bioinformatics analyses with minimal programming effort.
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34.
- Kuipers EJ, Grady WM, Lieberman D, Seufferlein T, Sung JJ, Boelens PG, van de Velde CJ, Watanabe T. Colorectal cancer. Nat Rev Dis Primers. 2015;1:15065.
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330-337.
- Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2013;41
- Hosseini ST, Nemati F. Identification of GUCA2A and COL3A1 as prognostic biomarkers in colorectal cancer by integrating analysis of RNA-Seq data and qRT-PCR validation. Sci Rep. 2023;13(1):17086. Link
- Zhang N, Zhang R, Zou K, Yu W, Guo W, Gao Y, Li J, Li M, Tai Y, Huang W et al: Keratin 23 promotes telomerase reverse transcriptase expression and human colorectal cancer growth. Cell Death Dis 2017, 8(7):e2961.
- Fonseca AS, Ramao A, Burger MC, de Souza JES, Zanette DL, de Molfetta GA, de Araujo LF, de Barros ELBR, Aguiar GM, Placa JR et al: ETV4 plays a role on the primary events during the adenoma-adenocarcinoma progression in colorectal cancer. BMC Cancer 2021, 21(1):207.
- Yao D, Bao Z, Qian X, Yang Y, Mao Z: ETV4 transcriptionally activates HES1 and promotes Stat3 phosphorylation to promote malignant behaviors of colon adenocarcinoma. Cell Biol Int 2021, 45(10):2129-2139.
- JEONG D, Ban S, Kim H, Oh S, Ji S, Kim HJ, Bae SB, Ahn TS, Kim C-J, Lee MS et al: Abstract 4455: Identification of novel oncogene, copine-7 (CPNE7), in colorectal cancer. Cancer Research 2017, 77(13_Supplement):4455-4455.
- Ferguson HJ, Wragg JW, Ward S, Heath VL, Ismail T, Bicknell R: Glutamate dependent NMDA receptor 2D is a novel angiogenic tumour endothelial marker in colorectal cancer. Oncotarget 2016, 7(15):20440-20454.
- Wang Z, Xia B, Qi S, Zhang X, Zhang X, Li Y, Wang H, Zhang M, Zhao Z, Kerr D et al: Bestrophin-4 relays Hes4 and interacts with Twist1 to suppress epithelial-to-mesenchymal transition in colorectal cancer cells. In.: Cold Spring Harbor Laboratory; 2023.
- Karunanithi S, Levi L, DeVecchio J, Karagkounis G, Reizes O, Lathia JD, Kalady MF, Noy N: RBP4-STRA6 Pathway Drives Cancer Stem Cell Maintenance and Mediates High-Fat Diet-Induced Colon Carcinogenesis. Stem Cell Reports 2017, 9(2):438-450.
- Kumara H, Bellini GA, Caballero OL, Herath SAC, Su T, Ahmed A, Njoh L, Cekic V, Whelan RL: P-Cadherin (CDH3) is overexpressed in colorectal tumors and has potential as a serum marker for colorectal cancer monitoring. Oncoscience 2017, 4(9-10):139-147.
- Hibi K, Goto T, Mizukami H, Kitamura YH, Sakuraba K, Sakata M, Saito M, Ishibashi K, Kigawa G, Nemoto H et al: Demethylation of the CDH3 gene is frequently detected in advanced colorectal cancer. Anticancer Res 2009, 29(6):2215-2217.
- Christodoulou S, Alexopoulou DK, Kontos CK, Scorilas A, Papadopoulos IN: Kallikrein-related peptidase-6 (KLK6) mRNA expression is an independent prognostic tissue biomarker of poor disease-free and overall survival in colorectal adenocarcinoma. Tumour Biol 2014, 35(5):4673-4685.
- Henkhaus RS, Gerner EW, Ignatenko NA: Kallikrein 6 is a mediator of K-RAS-dependent migration of colon carcinoma cells. Biol Chem 2008, 389(6):757-764.
- Dietinger V, Garcia de Durango CR, Wiechmann S, Boos SL, Michl M, Neumann J, Hermeking H, Kuster B, Jung P: Wnt-driven LARGE2 mediates laminin-adhesive O-glycosylation in human colonic epithelial cells and colorectal cancer. Cell Commun Signal 2020, 18(1):102.
- Kitamura T, Biyajima K, Aoki M, Oshima M, Taketo MM. Matrix metalloproteinase 7 is required for tumor formation, but dispensable for invasion and fibrosis in SMAD4-deficient intestinal adenocarcinomas. Lab Invest. 2009;89(1):98-105.
- Almendro V, Ametller E, Garcia-Recio S, Collazo O, Casas I, Auge JM, Maurel J, Gascon P. The role of MMP7 and its cross-talk with the FAS/FASL system during the acquisition of chemoresistance to oxaliplatin. PLoS One. 2009;4(3)